Die passende Antwort auf häufig gestellte Fragen
Finden Sie erklärende Informationen zu unseren Produkten und Dienstleistungen, indem Sie auf die folgenden Links klicken.
Allgemeine Fragen zur Probenübermittlung, Bestellung, zum Probenversand und zu Ergebnissen finden Sie hier:
Was soll ich tun, wenn meine Probe die Eingangskontrolle (Entry QC) nicht besteht?
Wenn die Menge, Konzentration oder Qualität der sequenzierbereiten Bibliothek nicht den Anforderungen für die weitere Probenverarbeitung entspricht, werden wir Sie kontaktieren, um das weitere Vorgehen im Projekt zu besprechen. Wann immer möglich, wird Eurofins Genomics Empfehlungen zur Optimierung der Probenqualität geben.
Ist das Sortieren von gemultiplexten Sequenzen inbegriffen?
Die Sortierung der Rohdaten anhand des Illumina-Index-Reads ist kostenlos. Der verwendete Indextyp (einfach indiziert oder (eindeutig) doppelt indiziert) sowie die entsprechenden Indexsequenzen müssen vor Projektbeginn im Proben-Einreichungsformular angegeben werden.
Garantieren Sie eine bestimmte Anzahl an Reads?
Die tatsächlich erzielte Sequenzierleistung (Read-Länge, Read-Qualität und Anzahl der Reads) hängt direkt von der Qualität der verwendeten Sequenzierbibliothek ab. Eurofins Genomics kann keine Garantie für Sequenzierdaten übernehmen, die aus vom Kunden vorbereiteten einsatzbereiten Bibliotheken stammen. Sollten unzufriedenstellende Sequenzierergebnisse auf technische Probleme mit den Sequenzierkits oder -geräten zurückzuführen sein, wird die Sequenzierung ohne zusätzliche Kosten für den Kunden wiederholt.
Kann Eurofins das Demultiplexing von „In-Line“-Barcodes in Sequenzier-Reads durchführen?
Ja, wir bieten das Demultiplexing von „In-Line“-Barcodes in Sequenzier-Reads (nicht in Index-Reads) an. Wir empfehlen jedoch dringend, auf die Verwendung von „In-Line“-Barcodes zu verzichten und stattdessen das Illumina-Indexsystem zu nutzen (d. h. die Barcodes werden in einem separaten Read ausgelesen und beeinträchtigen nicht die Cluster-Erkennung).
Es ist wichtig, darauf zu achten, dass die Basenzusammensetzung der Indizes ausgewogen ist, um die Signalunterscheidung durch die Bildanalyse-Software zu optimieren.
Bitte kontaktieren Sie uns für weitere Details. Für diesen Service fallen zusätzliche Kosten an.
Welche Art von einsatzbereitem Bibliothekspool kann ich einreichen?
Sie können jeden Bibliothekspool einreichen, der mit Illumina®-Sequenzierung kompatibel ist, einschließlich solcher, die aus genomischer DNA für Whole-Genome-Sequenzierung, RNA für Transkriptomanalysen oder Amplicon-Bibliotheken hergestellt wurden. Bitte beachten Sie, dass das Pooling verschiedener Bibliotheken zu einem finalen einsatzbereiten Pool von den Kunden selbst durchgeführt werden muss – dieser Service wird von Eurofins Genomics nicht angeboten.
Illumina-kompatible Bibliotheken müssen vier zentrale Komponenten in ihren Adaptersequenzen enthalten – diese befinden sich an den Enden jedes Fragments – um eine erfolgreiche Paired-End-Sequenzierung zu ermöglichen:
- Flow-Cell-Bindungsstellen (P5 und P7): Ermöglichen die Bindung und Clusterbildung der DNA-Fragmente auf der Flow Cell.
- Sequenzierprimer-Bindungsstellen (Read 1 und Read 2): Starten die Sequenzierung von beiden Enden des DNA-Fragments.
- Indexsequenzen (i5 und i7): Auch als Barcodes bekannt, ermöglichen sie das Multiplexing von Proben. Bibliotheken können einfach (ein Index) oder doppelt (zwei Indizes) indiziert sein. Für NovaSeq liegt die empfohlene Barcodelänge bei 8–12 bp, für MiSeq empfehlen wir maximal 10 bp lange Indexsequenzen.
- Indexprimer-Bindungsstellen: Diese sind notwendig, um die Indexsequenzen während der Sequenzierung auszulesen.
Single-Index Bibliothek
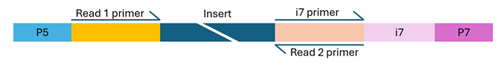
Dual-Index Bibliothek
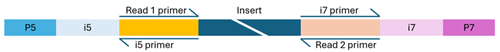
Die Struktur der Adaptersequenzen variiert je nachdem, ob die Bibliothek mit einfacher oder doppelter Indizierung arbeitet. Fehlende, falsch konfigurierte oder stark modifizierte Komponenten können dazu führen, dass die Sequenzierung fehlschlägt. Wenn Sie sich bezüglich der Konfiguration Ihrer Bibliothek unsicher sind, können Sie sich gerne an uns wenden – wir beraten Sie gerne.
Können Sie PhiX in meinen Sequenzierlauf einspiken?
Die Cluster-Erkennungsalgorithmen von Illumina sind auf eine ausgewogene Verteilung der Nukleotide A, T, G und C optimiert. Jede Abweichung von dieser Gleichverteilung wirkt sich negativ auf die Menge und Qualität der erzeugten Sequenzierdaten aus.
Um die Nukleotidverteilung in der Bibliothek auszugleichen, wird bei Proben mit geringer Diversität oder unausgewogener Basenzusammensetzung (z. B. Amplicons, bisulfitkonvertierte Proben) ein Spike-in von 20 % PhiX (bei MiSeq) verwendet.
Das Ausmaß, in dem der negative Einfluss einer unausgewogenen Basenzusammensetzung durch den PhiX-Kontroll-Spike-in reduziert werden kann, hängt von den individuellen Eigenschaften der Probe und der Sequenz ab.
Weitere Informationen finden Sie auf der Website von Illumina.
Können Sie eine Datenanalyse für mein einsatzbereites Projekt durchführen?
Ja, wir bieten auf Wunsch eine optionale Datenanalyse an. Bitte geben Sie dies im Angebotsformular an.